WP05 - Complement Disorders
Objectives
- To identify genetic and acquired initiators and modifiers of aHUS, MPGNI, DDD, and C3GN
- To develop technologies for diagnosis and screening of patients
- To find out disease-specific deep transcriptomic and proteomic profiles
- To discover biomarkers predicting response to therapies and post-transplant disease recurrence
- To develop in vitro, ex vivo and in vivo models for testing preventive and therapeutic interventions
Workpackage Description
Atypical haemolytic uremic syndrome (aHUS), primary membranoproliferative glomerulonephritis type I (MPGN I), dense deposit disease, (DDD) and C3-glomerulonephritis (C3GN) are disorders characterized by excessive activation of the alternative complement pathway (AP). HUS is a rare disease of microangiopathic haemolysis, thrombocytopenia and renal failure. The most common form, associated with infection by shiga-toxin producing Escherichia coli, has a good prognosis. The rarer atypical form (aHUS) has a poorer outcome, with 10-15% acute mortality and 50% of cases progressing to ESRD. Sixty percent of cases are related to mutations in genes encoding complement regulatory proteins, factor H (CFH), membrane-cofactor protein (MCP), factor I (CFI) and thrombomodulin (THBD), or the alternative pathway (AP) C3 convertase components C3 and factor B (FB). Hybrid CFH-CFHR1 genes have been reported. Homozygous CFHR1-CFHR3 deletion is associated with an autoimmune form with inhibitory anti-CFH autoantibodies. Such abnormalities cause complement hyperactivation in particular on the surface of microvascular endothelium and platelets leading to endothelial injury and thrombus formation in the kidney and in other organs. However, not all mutation carriers develop the disease. Multiple gene mutations/variants and environmental triggers may be required for disease manifestation. The type of genetic defect strongly impacts on outcome and recurrence rate after renal transplantation. Major therapeutic progress has been made recently with the discovery of Eculizumab, an anti-C5 antibody as effective treatment of aHUS.

- Figure 1: aHUS: a disease of complement hyperactivation on endothelial cells
MPGN I, DDD and C3GN are characterized by glomerular inflammation, proteinuria and progressive renal failure. Affected children develop end stage renal disease during late childhood or adolescence. These conditions are characterized by glomerular C3 deposition. C3NeF, an autoantibody stabilizing the AP C3-convertase, is found in 50-75% of patients. Auto-antibodies against CFH and factor B and complement gene mutations have been also reported. There is no specific treatment. Eculizumab reduced proteinuria in a few patients with MPGN I and DDD, but others did not respond. Several crucial issues remain unsolved that ask for innovative and integrated studies. The underlying cause has not been identified in about 40% of aHUS and 70% of MPGN I, DDD and C3GN patients. The formation of anti-CFH antibodies cannot be attributed only to CFHR1/CFHR3 deletion since the deletion is also present in 5% of normal individuals. The origin, mechanism of action and role of C3NeF are still unclear. The incomplete penetrance in mutation carriers and the highly heterogeneous phenotypic expression (patients with the same mutation may develop either aHUS, MPGN I, DDD or C3GN) indicate that multiple predisposing factors are involved and define the need to establish disease-specific genetic/molecular/proteomic signatures.

- Figure 2: Complement Activation Pathways
We have funded a European working party for the study of complement genetics in renal diseases which forms the core research teams involved in work package-5. The consortium, constituted of Italian, UK, Spanish, French and German teams has collected national and international cohorts of >1,500 patients with aHUS, MPGN I, DDD and C3GN with large biorepositories of serum and DNA from these patients and large collections of clinical data.
We will expand our resources by enrolling new patients. Particular emphasis will be put on a careful follow-up of patients.
Methodologies
Search for genetic initiators of complement-related disorders
In patients without identified abnormalities, exome/whole genome sequencing will be used to identify new disease-associated genes. Candidate gene variants will be validated by sequencing controls matched for ethnicity. New CFH/CFHRs gene rearrangements/deletions will be searched by MLPA, array CGH and sequencing Cell and tissue localization of novel gene products and the effect of mutations will be assessed in kidney biopsies from patients and controls. Functional studies and protein-protein interaction assays will be carried out using recombinant proteins or proteins isolated from patients. For in vivo studies and drug discovery purposes Xenopus morpholino knockdown and KI/KO mouse models of novel disease-associated genes will be studied (if already available) or generated de novo.

- Figure 3: Electron microscopy image of glomerular lesions in a patient with MPGN
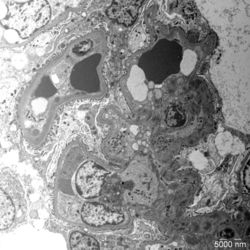
- Figure 4: Electron microscopy image of glomerular lesions in a patient with C3 glomerulopathy
Search determinants of disease penetrance and clinical heterogeneity in mutation carriers
Genetic determinants of disease penetrance will be investigated by exome sequencing of probands and unaffected mutation carriers. We will also identify complement quantitative trait loci by linkage analysis. Epigenetic determinants of disease penetrance will be also investigated.
Determinants of anti-CFH Abs formation and characterization of C3NeF
Genetic variants that synergize with CFHR1 deletion in predisposing to development of anti-CFH Abs, will be searched by exome sequencing of cases and controls. Mapping of the epitope(s) in the C3bBb C3 convertase recognized by C3NeF isolated from patients will be done.
Search for novel autoimmune causes of aHUS, MPGN I, DDD or C3GN
This task will be carried out by testing sera from patients for IgG binding to complement components or regulators by ELISA or western blot. Epitope mapping and the effects of the newly discovered antibodies on complement activation/regulation will be investigated.
Development of tools for screening and diagnosis of aHUS, MPGNI, DDD and C3GN
We will design a diagnostic panel for targeted parallel sequencing of known and newly discovered genes on Next generation sequencing platforms. False negative and positive errors will be checked by testing the panel in patients previously screened by Sanger sequencing. We will then use the panel to screen patients without known gene abnormalities. An ELISA kit and an assay for rapid and reliable detection of anti-CFH Abs and C3NeF will be developed.
Establish disease-specific deep transcriptomic and proteomic profiles
We will obtain mRNA, and miRNA profiles in PBMC, urine sediment and plasma exosomes, and proteomic profiles in plasma and urine sediment of patients carrying mutations in different genes or no gene mutation.
Develop in vitro, ex vivo and in vivo models for testing new preventive and therapeutic interventions
In vitro and ex vivo tests will be developed to evaluate complement activation profile in patients before and during specific treatments, to find out biomarkers of disease activity and of response to treatment. The assays will be also used for testing the effect of new complement inhibitors on the activation of the alternative, classic and lectin pathways of complement both in fluid phase and at cell surface level. Either Xenopus knockdown models of mouse ko or knock-in models will be used for selection of compounds with complement inhibitory activity in vivo.
Participating Partners
Heidelberg University Medical CenterHeidelberg University Medical Center
Istituto di Recerche Farmacologiche Mario Negri
Agencia Estatal Consejo Superior de Investigaciones Cientificas
University of Newcastle upon Tyne
Assistance Publique - Hopitaux de Paris
Leipniz Institute for Natural Product Research - Hans-Knöll-Institute
Multiplicom Inc.
Philogen S.p.A
Philochem AG
Comprehensive Biomarker Center
mosaiques diagnostics GmbH
Ludwig-Maximilians-University Munich - Walter Brendel Zentrum für Experimentelle Medizin