University of Michigan

University of Michigan
560 MSRB II, 1150
W. Medical Center Dr.-SPC5676
48109 Ann Arbor
USA
Project Leader

Prof. Matthias Kretzler
Phone: + 1 734 615 5757
Fax: + 1 734 763 0982![]() Contact
Contact
Project Staff

Assistant Prof. Yuanfang Guan
Co-Investigator![]() Contact
Contact
Institute Presentation
In biomedicine we are currently experiencing a fundamental transition from research models focused on the functions of single molecules or pathways to an integrative biology analyzing biological systems as a unified whole. A comprehensive molecular analysis of the tissue and associating the expression signatures to the relevant clinical parameters will aid in predicting disease course and response to therapy. Systems biology approaches in medicine are expanding rapidly as novel technologies allow for analysis across genome-scale data sets to define key drivers of diseases. Studying the molecular networks in multiple relevant tissues and associating them with clinically relevant phenotype data and other genomic data to identify the networks driving disease is at the center of what has been loosely defined as systems biology.
To implement a systems biology approach, an interdisciplinary, international, multi-center approach integrating clinical and experimental nephrology, pathology, immunology, molecular biology, bioinformatics and epidemiology has been implemented by our team at the Applied Systems Biology Core at the University of Michigan over the past five years.
Our Systems Biology approach can be broadly divided into three main components (Fig1):
1) A highquality sample procurement core drawing from a wide spectrum of studies (EURenOmics, C-PROBE, Neptune)
2) An effective data analysis pipeline drawing on deep Bioinformatic expertise utilizing the latest technologies available for efficient data processing and mapping to generate large scale genomic data
3) Expert integration of these large multi-scalar genomic, clinical and histological data into their biological functional context of specific kidney diseases by domain experts of clinical investigators utilizing several approaches of network/pathway/visualization tools
With this infrastructure and wide range of expertise in place, the molecular mechanism underlying rare renal diseases with an emphasis on identifying biomarkers of progression and therapeutic targets can now be addressed. EURenOmics is in the unique position to generate genome-scale molecular profiles along the genotype-phenotype continuum of a wide spectrum of disease. Establishing the dependencies between molecular data sets and comprehensive clinical profiles will allow the research partners to define kidney diseases using mechanistic rather than descriptive concepts. Subsequently, predictors of outcome and treatment response can be defined in functional terms and novel regulatory pathways identified for targeted intervention. Systems genetics approaches will be employed to define regulatory dependencies between genotypic risk, molecular profiles and phenotypic parameters. The combination of the EURenOmics investigators insight into specific research areas with the large-scale molecular datasets annotated to human disease plus the expertise in integrative data mining will generate novel hypothesis and insight into human renal disease.
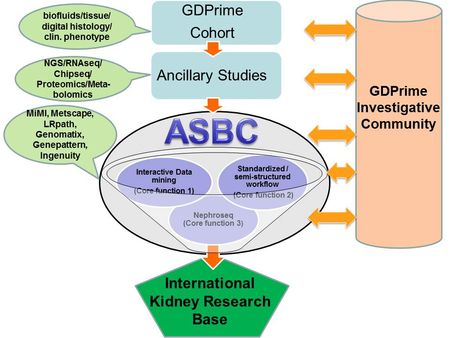
- Fig. 1: Workflow at the Applied Systems Biology Core (ASBC) at U. Michigan
Our translational systems biology research pipeline allows for optimal genome scale data analysis from genes to function. EURenOmics will provide a unique opportunity to analyze cohorts with prospective, well phenotyped cohort of patients with rare renal disease. Matching biosamples of kidney tissue, blood and urine will continue to be procured using Standard Operating Procedures. A large number of projects initiated by EURenomics investigators have already generated genome-scale data sets. The European Renal cDNA Bank (ERCB) has provided samples for large-scale analyses by the international renal research community. We have performed expression profiles from micro-dissected renal tissues from human and murine samples and have generated comprehensive genome-wide data sets from glomerular and tubulo-interstitial compartments from a wide spectrum of rare renal diseases and control samples. We also have generated high throughput RNA-seq transcriptomics data, metabolomics data and whole-genome SNP data for a subset of these samples. This extensive collection of expression profiles along with high quality well documented morphometric and longitudinal clinical data will serve as the basis for our research.
Using standardized and semi-structured workflows involving GenePattern (Fig.2), we ensure highly efficient and transparent data pre-processing, quality control and data-integration.
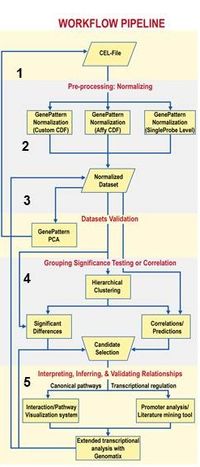
- Fig. 2: Workflow to guide identifying genes of interest from high throughput analysis.
Multi-scalar data integration: EURenOmics is specifically designed to enable generation of multiple layers of genome wide data in the same individual with kidney disease. Our SME bioinformatics partner Genomatix (www.genomatix.com) has established in close collaboration with us an analysis pipeline capable of combining diverse genome wide data sets for comprehensive molecular disease definitions. Fig. 3 summarizes the data analysis and mining pipeline. Integrating these data sets across the different genome scale data domains, referred to here as multi-scalar analysis, has the potential to identify key drivers of human disease. Such integration will move these analyses from the level of pure association to one of causal inference. Systems genetics approaches will be employed to define regulatory dependencies between genotypic risk, molecular profiles and phenotypic parameters.
These interactive complex data interaction demands the use of a web based interactive, shared data mining portal. The National Center for Integrative Bioinformatics (NCIBI) data portal (http://portal.ncibi.org/gateway/) combines the need of not only an effective communication and training platform but also solves the data storage requirements. A secure web portal allows investigator involved in the project to store and share data, manuscripts and presentations. Because the portal is a web-based platform for both collaboration and analysis, many of the limitations of locally managed data analysis solutions will not be applicable. The portal also serves as a suite of a multitude of bioinformatic analysis tools employed by the ASBC team to serve our distributed research team. The portal structure has proven to be essential for real-time online assistance through available diverse web based communication channels.
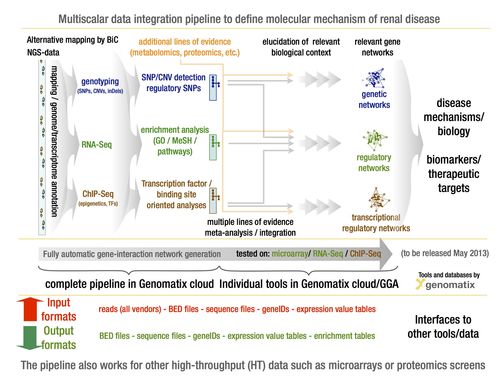
- Fig. 3: GENOMATIX multi-scalar data integration workflow to define specific and shared molecular pathways in rare renal disease
In order to facilitate the optimal usage of very rich renal data sets by worldwide kidney research teams we developed a web-based research interface for analyzing complex, disease-specific gene expression data sets using a predefined analysis algorithm. The Nephroseq system (www.nephroseq.org) is a kidney-specific version of the highly successful cancer-specific data-portal, Oncomine (www.oncomine.org) and has been developed in collaboration with Drs. Rhodes and Chinnaiyan. Nephroseq is a fully-automated web-based systems biology search engine for context specific renal disease gene expression data mining (Fig. 4). Nephroseq contains human renal gene expression data sets from nearly 2,000 samples encompassing over 59 million gene expression measurements across 15 chronic kidney diseases. Nephroseq is freely available to the worldwide academic community through a web-based interface that allows interrogation of the data sets with two distinct tools. First, Nephroseq allows retrieval of disease and tissue-specific gene expression values for defined molecules of interest. Second, it enables advanced analysis of entire gene expression data sets for a given disease entity by linking to system biology tools in a predefined automated manner. Nephroseq computes differential expression using uniform analysis tools; performs meta-analysis of gene expression across studies including concept analysis using gene set enrichment tools; and displays molecular interaction of co-regulated genes. The automated integration of different external resources into the data warehouse, working in the software background (Fig. 4), allows user-friendly data export and analysis covering a broad spectrum of functionalities. Data sources in Nephroseq include those from the public domain (GEO at NCBI and Array Express at EBI), studies generated in the framework of the OBrien Renal Center, and data sets obtained by request from ongoing or published studies. Data generated in the UM OBrien Renal Center are often placed into Nephroseq immediately and no later than 3 years after generation. Nephroseq has been rapidly adopted by kidney researchers.
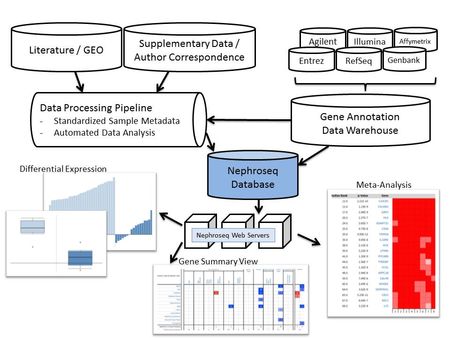
- Fig. 4: Structure and capabilities of Nephroseq. The database consists of three layers: data input, data analysis, and data visualization. The data input layer has two components, the data processing pipeline and the gene annotation data warehouse. The expression pipeline is used internally to identify and prioritize studies in the literature. The pipeline also draws data directly from public resources. The data-analysis layer consists of sample metadata standardization and automated statistical analysis. The sample metadata standardization utilizes the NCI Thesaurus and manual annotation. The automated statistical analysis component is implemented in Perl and R. A series of scripts monitor the database for new data and sample parameters and automatically performs differential expression analysis, cluster analysis, and concept analysis, when needed. The Nephroseq web servers query data from the Nephroseq database and display tabular and graphical representations of the data and analysis results.
Expertise of key personnel
Matthias Kretzler, MD has more than 14 years of experience in shared integrated data mining of molecular renal data sets. He has conceived and implemented the most comprehensive multi-center, international study for gene expression of renal disease, the European Renal cDNA Bank (ERCB), he has lead the ASBC over the past five years, has initiated the Nephrotic Syndrome Study Network for integrated systems biology of glomerular diseases. He recruited and trained a research team for kidney disease focused gene expression data mining. He integrated the kidney-specific research focus into the unique bioinformatics environment at the University of Michigan, drawing on the extensive expertise in bioinformatics database management, genome-wide expression data set analysis and data mining using a wide spectrum of approaches available in CCMB and NCIBI. His systems biology research focus is on molecular marker definition of renal disease and definition of transcriptional networks in renal disease. In addition, he has long standing expertise in podocyte cell biology. As a faculty member in the Department for Computational Medicine and Biology he is responsible for the systems biology segment of the Introduction to Bioinformatics and Computational Biology course in the University of Michigan Bioinformatics doctoral program.
Dr. Kretzler will be responsible for the coordination of the diverse aspects of the bioinformatics data integration in EURenOmics. He will use his extensive experience integrating bioinformatics, molecular and clinical approaches, which served more than 70 collaborative research teams, to maintain a standardized data processing pipeline, continuously update the available data mining tools, and ensure optimal applicability of the research tools for the goals of the EURenOmics network.
Yuanfan Guan, PhD, Assistant Professor, Department of Computational Medicine and Bioinformatics and Internal Medicine/Nephrology, Co-I (1.2 month effort): Dr. Guans long-standing research interest is to develop bioinformatics tools for integrating heterogeneous functional genomics data, which can assist to understand the protein functions and molecular mechanisms behind renal diseases. She develops statistical and machine learning tools for inferring protein functions and phenotypes though heterogeneous data integration (Guan et al., 2008, Genome Biology, Guan et al., 2010, PLoS Comp. Biol.; Guan et al., in press, PLoS Comp. Biol). The software and tools developed (mouseNET, mouseMAP, KOMPute) are intensively used by the genetics community.
Since her recruitment to U-M in 2011 with joint appointment in the Division of Nephrology and the Department of Computational Medicine and Bioinformatics, she has focused on predicting activated pathway in renal disease, modeling expression changes across disease course, integrating context-specific networks relevant to kidney diseases and predicting isoform functions related to genes causing kidney diseases.
She will use her expertise in the ASBC to adapt her previous tools for genome-wide functional genomic data integration to renal disease-specific data integration and mining. To this end, she has established a locally maintained cluster that has the capability to process all publicly available functional genomic data (including RNAseq) and the forthcoming renal-specific functional genomic data generated by the network. She will apply the Bayesian network approach developed previously for global and tissue-specific networks to generate renal disease-specific networks. She will employ an algorithm prioritizing disease-associated genes and pathway components through mining large-scale, heterogeneous datasets in the ASBC for functional prediction and pathway association of novel disease genes. In addition, she will be responsible for functional analysis of transcripts identified through RNAseq and proteomic data to associate them with the potential roles in renal diseases. Finally, she will be responsible for integrating the above machine learning and statistical tools into user-friendly interfaces in the context of Nephroseq.